生信步骤|MAFFT结合HMMER进行多序列比对和基于隐马模型的基因搜索 |
您所在的位置:网站首页 › hmmer 软件使用 › 生信步骤|MAFFT结合HMMER进行多序列比对和基于隐马模型的基因搜索 |
生信步骤|MAFFT结合HMMER进行多序列比对和基于隐马模型的基因搜索
|
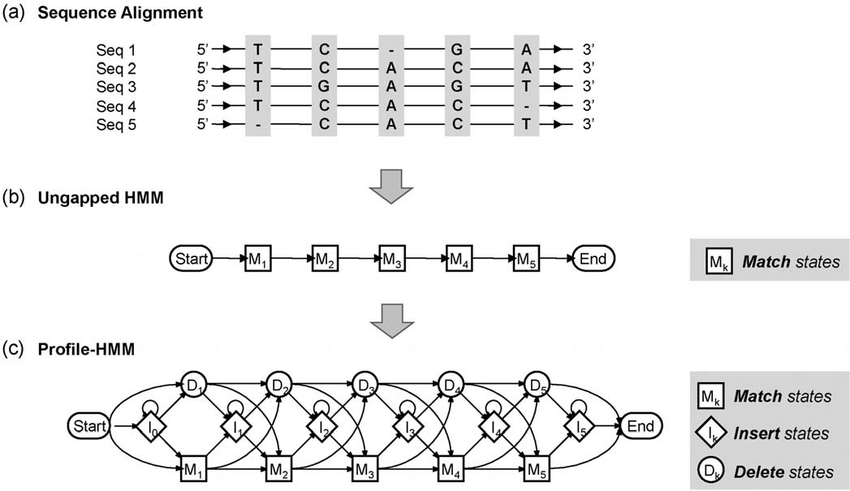
蛋白质都是由相似的小型结构域组成的。如果我们有若干个已知的蛋白序列,那我们就可以根据这些蛋白序列比较其含有的保守域,寻找在蛋白数据库中上是否也有一样保守域的蛋白。而后根据统计学模型,将显著性较高的蛋白序列预测为同一类基因家族蛋白。 随着蛋白质数据库的日趋完善,使用蛋白质结构域进行序列比对相比起传统的序列全长比对更具优势。对于每个蛋白质家族,通常有数千个已知的同源蛋白可以比对成较深的多重序列。序列比对揭示了一种特定于该结构域的结构和功能的进化模式(profile)。这些模式可以用概率模型捕捉到。HMMER能够从蛋白或核酸序列中提取出域家族从而构建隐式马尔可夫模型(profile hidden Markov models, profile HMMs),从而用于同源序列检索,注释新的序列。 需要用到的软件包括mafft,hmmer,seqkit。 $ conda install -c bioconda mafft hmmer seqkit 2.MAFFT多序列比对蛋白质MAFFT是一款多序列比对软件,相比起多序列比对的明星软件ClustalW,MAFFT在准确性和速度上均具有优势。准确性上MAFFT>Muscle>T-Coffee>ClustalW,比对速度上Muscle>MAFFT>ClustalW>T-Coffee [1]。因此我们在这里采用MAFFT进行多序列比对。 将待比对的序列手动收集并存放于ZAR1_new.fas文件后,我们采用MAFFT的方式进行多序列比对。注意:mafft可调整的参数较多,可根据需求选择适当的参数。 $ mafft --localpair --maxiterate 1000 ZAR1_new.fas > ZAR1_aligned.fas 2.1可调整的比对算法:2.1.1 mafft准确度优先的比对算法(Accuracy-oriented methods): #L-INS-i (最准确的算法;适用于200条序列以下的比对): $ mafft --localpair --maxiterate 1000 input [> output] #G-INS-i (适用于序列长度相似的比对;200条序列以下为佳): $ mafft --globalpair --maxiterate 1000 input [> output] #E-INS-i (适用于包含大范围非比对区的序列;200条序列以下为佳;--ep 0选项代表允许超长gap的出现): $ mafft --ep 0 --genafpair --maxiterate 1000 input [> output]2.1.2 mafft速度优先的比对算法(Speed-oriented methods): #FFT-NS-i法: $ mafft --retree 2 --maxiterate 2 input [> output] #FFT-NS-i法(最高1000次迭代): $ mafft --retree 2 --maxiterate 1000 input [> output] #FFT-NS-2法(快): $ mafft --retree 2 --maxiterate 0 input [> output] #NW-NS-2法(快,且不进行FFT近似估计): $ mafft --retree 2 --maxiterate 0 --nofft input [> output] #FFT-NS-1法(更快; 推荐在比对2000条以上序列时使用): $ mafft --retree 1 --maxiterate 0 input [> output] #NW-NS-PartTree-1 (推荐序列数~10,000到~50,000条时使用): $ mafft --retree 1 --maxiterate 0 --nofft --parttree input [> output]2.1.3 mafft群体间比对的算法(Group-to-group alignments): $ mafft-profile group1 group2 [> output]以上MAFFT的参数命令行均有简写形式,详情请见Mafft Manual 。 3.hmmbuild将多序列比对文件转化为隐马模型我们采用HMMER软件进行隐马模型的建立。 #将多序列比对文件转化为隐式马尔可夫模型 $ hmmbuild ZAR1.hmm ZAR1_aligned.fas 4.利用隐马模型搜索结构域类似的蛋白质通过隐马模型搜索蛋白数据库中符合该结构的蛋白质。将刚产生的profile轮廓文件作为输入,检索靶向数据库中符合该轮廓的蛋白序列,最终按照符合度输出序列结果。Hongyang_pep.fa是先行下载好的蛋白质组序列文件,以fasta格式呈现。 #在Hongyang_pep.fa蛋白质组中搜索具有ZAR1.hmm特征的蛋白质 $ hmmsearch ZAR1.hmm Hongyang_pep.fa > hmmer_result.out #设定bit-score阈值筛选搜索结果,此处设定bit-score阈值为15 $ hmmsearch -T 15 ZAR1.hmm Hongyang_pep.fa > hmmer_bit.out #设定bit-score阈值筛选搜索结果。默认为 10, 表示每个搜索报告大约 10 个错误结果。 $ hmmsearch -E 0.0001 ZAR1.hmm Hongyang_pep.fa > hmmer_e.out #此处设定过滤阈值-E如果是e-100类似形式会报错,因此建议比对后使用awk进行过滤。此外,常用到的HMMER命令还包括: hmmbuild: 用多重比对序列构建HMM模型; hmmsearch: 使用HMM模型搜索序列库; hmmscan: 使用序列搜索HMM库; hmmalign: 使用HMM为线索,构建多重比对序列; 5.获取匹配的蛋白质并进行tblastn检索新的同源蛋白利用hmm模型在蛋白质组序列中寻找相似的蛋白后,可以通过seqkit提取该序列(新建grep.txt文件并将待提取序列的名称保存于此)。再通过tblastn会将库中的核酸翻译成蛋白序列,在核酸库中寻找与该蛋白相似的核酸序列。实际使用时可以采用全基因组建立核酸库,即可搜索全基因组内可能与目标蛋白相似的序列。 #seqkit提取隐马模型预测的序列,保存于Actinidia05846.t1.fa文件 $ seqkit grep -f grep.txt Hongyang_pep.fa -o Actinidia05846.t1.fa #基因组建立核酸库并命名为:canu_genome $ makeblastdb -in 11251AaHscanu.contigs.fasta -dbtype nucl -parse_seqids -input_type fasta -out canu_genome #核酸库中比对目标序列 $ tblastn -db canu_genome -query Actinidia05846.t1.fa -outfmt 6 -out tblastn_canu.result 6.对新检索的蛋白序列进行HMM结构域的注释对于刚刚找到的蛋白,如果我们希望探究其功能,往往会对其结构域进行搜索。毕竟结构决定了功能。待探究的输入文件可以是单个蛋白序列,多蛋白序列,hmm隐马模型。我们采用pfam网站对蛋白序列进行注释。首先需要下载Pfam注释库文件,Pfam网站中保留的库文件目前只有A数据库,A数据库代表着经过手工校正的高质量数据库。此外,我们还需要对库文件进行初步的二进制压缩和引索处理。 #下载Pfam库文件 $ wget https://ftp.ebi.ac.uk/pub/databases/Pfam/current_release/Pfam-A.hmm.gz #解压Pfam库文件 $ gzip -d Pfam-A.hmm.gz #压缩Pfam库文件:此处的Hmm文件以文本形式保存,压缩为二进制有助于加速运算,建立成索引数据库。 $ hmmpress Pfam-A.hmm而后我们需要用到HMMER软件中的hmmscan进行Pfam注释。将待比对的序列放在hongyang_RPM1_like.fa文件中,使用刚刚压缩得到的库Pfam-A.hmm作为注释参考。得到的三个文件result.txt,result.tbl,result.dom分别表示待比对序列的注释结果文件,注释出的蛋白域信息文件,带有起止位置信息的蛋白结构域信息文件。 #hmmscan搜索蛋白质所含的结构域 $ hmmscan -o result.txt --tblout result.tbl --domtblout result.dom --noali -E 1e-5 Pfam-A.hmm hongyang_RPM1_like.fa #更多可选参数: # -h:显示帮助信息 # -o FILE:将结果输出到指定的文件中。默认是输出到标准输出。 # --tblout FILE:将蛋白质家族的结果以表格形式输出到指定的文件中。默认不输出该文件。 # --domtblout FILE:将蛋白结构域的比对结果以表格形式输出到指定的文件中。默认不输出该文件。该表格中包含query序列起始结束位点与目标序列起始结束位点的匹配信息。 # --acc:在输出结果中包含 PF 的编号,默认是蛋白质家族的名称。 # --noali:在输出结果中不包含比对信息。输出文件的大小则会更小。 # -E FLOAT:设定 E_value 阈值,推荐设置为 1e-5 。 # -T FLOAT:设定 Score 阈值。 # --domE FLOAT:设定domain比对的E_value阈值。类似-E参数。 # --cpu:多线程运行的CPU。默认应该是大于1的,表示支持多线程运行。但其实估计一般一个hmmscan程序利用150%个CPU。并且若进行并行化调用hmmscan,当并行数高于4的时候,会报错:Fatal exception (source file esl_threads.c, line 129)。这时,设置--cpu的值为1即可。结果文件示例如下: Query: Actinidia05846.t1 [L=955] Scores for complete sequence (score includes all domains): — full sequence — — best 1 domain — -#dom- E-value score bias E-value score bias exp N Model Description ------- ------ ----- ------- ------ ----- ---- – -------- ----------- 1.1e-60 205.0 0.2 1.7e-60 204.5 0.2 1.3 1 NB-ARC NB-ARC domain 7.6e-24 83.9 0.2 2.9e-23 82.1 0.2 2.1 1 Rx_N Rx N-terminal domain 3.4e-06 26.8 16.8 0.0049 16.7 0.5 4.6 5 LRR_8 Leucine rich repeat 参考资料: 多序列比对算法MAFFT以及HMMER和profile文件的使用 CSDN:https://blog.csdn.net/weixin_45429249/article/details/109021162HMMER User’s Guide. http://eddylab.org/software/hmmer/Userguide.pdfMafft Manual. https://mafft.cbrc.jp/alignment/software/manual/manual.htmlHMMSCAN使用pfam数据库对多序列文件进行结构域注释。https://www.jianshu.com/p/f6db8af1e2cb |
【本文地址】
今日新闻 |
推荐新闻 |